Синдром на ловец

Съдържание
синдром на Хънтър

синдром на Хънтър (мукополизахаридоза Тип II) - рядко генетично заболяване, съединявани с Х-хромозомата, където недостатъчност се дължи на частично ензимно разграждане и натрупване на киселинни мукополизахариди (гликозаминогликани) в различни тъкани. Честотата на деца, родени със синдрома на Хънтър е около 1: 100,000-150,000 новородените. В момента в света има не повече от 2000 пациенти с синдрома на Hantera- руски официални статистически данни не са на разположение. синдром на Хънтър се отнася към групата на безстопанствени заболявания, чието лечение, съгласно действащото законодателство, трябва да се финансира от федералните и регионалните бюджети.
Причини за синдром на Хънтър
синдром на Хънтър развитие е свързано с мутация на ген iduronatsulfatazy (IDS), кодираща лизозомна ензимът е идуронат-2-sulphatase. IDS ген е свързан с локус Xq28 на дългото рамо на X-hromosomy- понастоящем известни повече от 150 различни мутации ИТ (точкови мутации, малка и голяма делеция, вмъкване, настройка и така нататък.).
Поради свързан наследство Х-свързана, синдром на Хънтър обикновено засяга само момчета (XY). Хетерозиготни жените в повечето случаи са тип II мукополизахаридоза носители без клинични прояви, но някои случаи са описани синдром на Хънтър при момичета, свързани с нова мутация или инактивиране втория нормално X хромозома.
Мутации в гена IDS са придружени с дефицит или отсъствие на ензим идуронат-2-сулфатаза (I2S), непълно разцепване и натрупването в лизозомите на клетки от почти всички тъкани и органи на гликозаминогликани (GAGs) - дерматан сулфат и хепаран сулфат.
Класификация на Хънтър Синдром
Тежестта на клинични прояви синдром Hunter (средата на тегло, теглото) зависи от вида на мутация в гена IDS. Например, големи делеции и комплекс преструктуриране, като правило, са отговорни за развитието на тежки форми на синдрома на Хънтър. За тежка (мукополизахаридоза тип IIA) се характеризира с ранно развитие на клиничните симптоми (предимно на възраст 18-36 месеца), бързо прогресиращ разбира се, тежка умствена изостаналост, полиорганна недостатъчност. Пациенти с тежка форма на синдром на Хънтър умират през второто десетилетие от живота.
Умерена до образуване (мукополизахаридоза тип IIB) е приблизително 1/3 от всички патология. Клиничните прояви на синдрома на Хънтър обикновено се появяват при деца на 3-8 (понякога 10-13) година-интелигентност обикновено се съхранява продължителността на живота при благоприятни условия може да достигне до 50-60 години. Пациентите с леко синдром на Хънтър успешно могат да се реализират в професионалната сфера и имат здрави деца.
Симптомите на синдрома на Хънтър
В момента на раждането на деца със синдром на Хънтър се появи клинично здрави. Основните симптоми, разработени средно 2-4 години. Преди тази възраст симптомите на заболяването са неспецифични: деца може да развиете повторни ринит, шумно дишане, пъпен и ингвинална херния, хидроцеле.
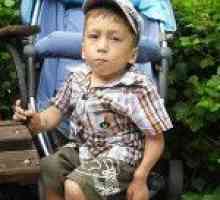
Рано характерна черта на синдрома на Хънтър е постепенна промяна във външния вид на детето: чертите на лицето стават груби (gargoilizm) - езика, устните и ноздрите - bolshimi- кожа - дебела. Виж пациент със синдром на Хънтър допълнена нанизъм, увеличаване на размера на главата (макроцефални), кратко гърлото, нарушено зъбите (зъби редки). Децата със синдром на Хънтър е външно много близки един до друг и напомнят на братята.
Други ранни признаци на мукополизахаридоза тип II включват дрезгав дълбок глас, коремни разширяването, хепатоспленомегалия. Децата със синдром на Хънтър са склонни към чести заболявания ТОРС, отит, ларингит, трахеит, пневмония- често те отпразнува обструктивна сънна апнея, хронична диария. В резултат на отлагане на гликозаминогликани и липиди в дермата е появата на нодуларно папулозен обрив по кожата на раменете, бедрата, лопатките. Може би появата на "монголски петна"В лумбосакралната област, хипертрихоза.
Още в възрастови деца в предучилищна възраст със синдром на Хънтър, има признаци на поражение на опорно-двигателния апарат води до скованост на ставите, развитие kyphoscoliosis, щам четки от типа "нокти". Движението става трудно, походка neuklyuzhey- за младежка възраст на пациентите със синдром на Хънтър често се прави безпомощни инвалиди, прикован към леглото.
Неврологични разстройства, възникващи в синдрома на Хънтър включват синдром свръхвъзбудимост, конвулсии, общуването хидроцефалия, спастична параплегия, забавено развитие на речта, прогресивен загуба на слуха. От визуална система се открива помътняване на роговицата, атипична ретинитис пигментоза. За най-скорошните признаци на тип мукополизахаридоза II включва сърдечни заболявания: появата на шум на сърцето, придобити сърдечни пороци (често митрална инсуфициенция) кардиомиопатия и др.
Детето може да изпита психични промени: обида, агресия. В зависимост от вида на синдрома на Хънтър интелектуална дефект може да бъде малко или много изразен.
Смъртен изход при пациенти със синдром на Хънтър обикновено се появява от прогресивна сърдечна или белодробна болест.
Диагностика на Хънтър Синдром
В педиатричната практика при синдром на Хънтър се случва много рядко. Въпреки това, може да се подозира, мукополизахаридоза тип II въз основа на клиничните признаци (външни промени, прояви на болестта в продължение на 2-4 години от живота, както и други прогресивни разбира се.). За да се потвърди диагнозата на деца, нуждаещи се от консултиране генетика, анализ на клинични и генеалогичните данни, извършване на молекулярно-генетични изследвания.
Важен биохимичен маркер синдром на Хънтър се увеличава екскрецията мукополизахариди (дерматан сулфат, хепаран сулфат) в урината, ензим дефицит идуронат-2-сулфатаза. Рентгенови изследвания на черепните кости, стави, дългите кости, гръбначния стълб показват дизостоза, остеоартрит, няколко промени на прешлените.
За да се проследят морфологичните и функционални промени на вътрешните органи и ЦНС извършва коремна ехография, електрокардиография, ехокардиография, електроенцефалография, мозък MRI. . Морфологично изследване на тъкани, получени чрез биопсия на кожата, черния дроб, миокарда и т.н., детектира промени от същия тип - клетки, пълни гликолипиди.
В семейства с известна генотип и висок риск от има дете със синдром на Хънтър може да се проведе инвазивна пренатална диагностика - хорион въси, амниоцентеза или cordocentesis с определянето на активността на идуронат-2-сулфатаза в получения материал.
Диференциална диагноза на синдрома на Хънтър трябва да се извърши с други форми на мукополизахаридоза (особено синдром на Hurler), както и други заболявания съхранение лизозомна.
Хънтър Синдром Лечение
Децата със синдром на Хънтър е необходимо да се проведе живота на ензим-заместваща терапия. Към днешна дата, единственото лекарство, регистрирано в САЩ, Европа и Русия за лечение на пациенти със синдром на Хънтър е Elapraza (Idursulfase). Пациенти с тип II мукополизахаридоза Elaprazu трябва да получават седмично чрез интравенозно вливане при доза от 0.5 мг / кг телесно тегло.
Освен патогенетичен лечение, деца със синдром на Хънтър, редовните цени на лечебни симптоматични и коригиращи терапия Хепатопротектори, витамини, антиоксиданти, cytoprotector. В комплекс лечение на синдрома на Хънтър е подходящо да се включат упражнения терапия, физиотерапия (електрофореза lidazy ставите, парафинови бани, магнитотерапия, лазерно пробиване), сесии с логопед и дефектология.
Прогнози и превенция на синдрома на Хънтър
Хънтър синдром тип Б е по-благоприятен курс и prognoz- с навременна и редовна лечение на пациенти с продължителност на живота може да бъде до 50-60 години. При тежки форми на мукополизахаридоза тип II пациентите обикновено умират преди 20 години сърдечно-съдови заболявания. Към днешна дата, е сериозен проблем за пациентите със синдром на Хънтър е навременно получаване на основни лекарства Elapraza поради високата си цена.
Децата със синдром на Хънтър трябва да наблюдават различни експерти: детски генетика, педиатър, детски кардиолог, невролог от децата, epileptologa, педиатрична офталмология, педиатрична Отоларинголог, детска хирургия, детската ортопедия.
Основният начин за предотвратяване на синдрома на Хънтър е генетично консултиране на двойки с вероятността за болно дете раждане, както и провеждане на пренаталното диагностициране. Трябва да знаете, че при пациенти със синдром на Хънтър се раждат здрави синове и дъщери служат obligantymi носители на мутантния ген.
 Мукополизахаридоза
Мукополизахаридоза Маточната-черепната синдром
Маточната-черепната синдром Синдром на Klippel Feil,
Синдром на Klippel Feil,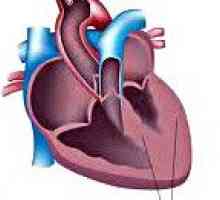 Вторичен кардиомиопатия
Вторичен кардиомиопатия Синдром на Goodpasture
Синдром на Goodpasture Синдром на Бартер
Синдром на Бартер Брахидактилия
Брахидактилия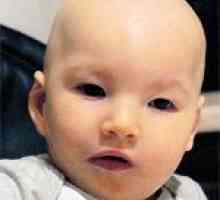 Синдром Cat вик
Синдром Cat вик Синдром на Ehlers - Danlos
Синдром на Ehlers - Danlos Синдром на Марфан
Синдром на Марфан Сколиоза при деца
Сколиоза при деца Синдром Shereshevscky, Turner
Синдром Shereshevscky, Turner- Загуба на слуха, увреждане на слуха
- Стволови клетки терапия
 Homogentisuria
Homogentisuria- Синдром на Търнър
 Синдром на Даун (тризомия 21)
Синдром на Даун (тризомия 21) Алпорт синдром
Алпорт синдром Синдром на Марфан
Синдром на Марфан Синдром Diamond shvahmana
Синдром Diamond shvahmana- Ябълкови чуплив Х-хромозома
 Синдром Cat вик
Синдром Cat вик Вторичен кардиомиопатия
Вторичен кардиомиопатия Алпорт синдром
Алпорт синдром Синдром на Даун (тризомия 21)
Синдром на Даун (тризомия 21) Мукополизахаридоза
Мукополизахаридоза Homogentisuria
Homogentisuria Синдром на Klippel Feil,
Синдром на Klippel Feil, Синдром Shereshevscky, Turner
Синдром Shereshevscky, Turner