Синдром на Klippel Feil,

синдром на Klippel Feil,
- генетично определена структурна аномалия на шийните прешлени, включително намаляване на броя и сливането на прешлените. Клинично определя визуално съкращаване на врата, ниско косата разположен в задната, ограничаване на движение на главата. Обикновено, синдром на Klippel-Feil е свързана с други вродени аномалии на скелетните и соматични органи. Диагнозата включва разнообразие от тесни специалисти, извършва рентгенов, CT и MRI на гръбначния стълб, генетичен анализ, разширено изследване на вътрешните органи (сърце, бъбреци, бели дробове, мозъка). Консервативно лечение се извършва с помощта на масаж, упражнения терапия и рехабилитация. Възможна хирургично лечение - хирургия tservikalizatsii.синдром на Klippel Feil,

синдром на Klippel-Feil - вродена, генетично обусловена патология на шийните прешлени, се състои в синтез на (синостоза) и намаляване на броя на прешлени. Най-често срещаният и постоянна характеристика на синдрома е значително съкращаване на шията, във връзка с които медицинската практика, също така е по-синдром къс врат. В повечето случаи в съчетание с други аномалии на системата на опорно-двигателния развитието (крива шия, сколиоза, болест Sprengel е, хипоплазия на горния крайник, синдактилия) И вродени вътрешни органи (бъбреци, сърдечно-съдовата система, белите дробове). синдром на Klippel-Feil е рядко заболяване. Честотата на възникването му - около 1 случай на 120 хиляди новородени .. За първи път този синдром е описан през 1812 г. във Франция от невролог и рентгенолог имена Klippel Feil, че в основата на неговото име.
Модерна клинична неврология класифицира Klippel-Feil синдром тип 3. Първият тип - KFS1 - се характеризира с намален брой на шийните прешлени. Обикновено човек в шийните прешлени 7, когато KFS1 обикновено 4-5. Вторият тип - KFS2 - синостозата на прешлени на шийните прешлени, и тяхното сближаване с тилната кост и гръдната прешлени. Третият тип - KFS3 - представлява комбинация от първия или втория прешлен на сближаването в долната гръдната и лумбалните прешлени. Често в района на шийката на матката, а има и допълнителни ребра спина бифида - разпукнат вертебрални арки.
Причини за синдром на Klippel-Feil
синдром на Klippel-Feil отнася до генетично определено заболяване. От време на време има спорадични случаи на синдрома. Аномалия образува утробата, все още в ембрионален период поради хипогликемия и аплазия, разделяне разстройства цервикални сегменти сливат забавяне по време Маркери прешлени. Генетични отклонения са хетерогенни. Когато те засягат KFS1 локус q22.1 на хромозома 8, при KFS2 са q22.1 локус на хромозома 5, с KFS3 - локус q13.31 12 хромозома. Най-проучен е GDF6 гена, отговорен за причиняване KFS1. Мутациите в този ген резултат в нарушение на синтеза на протеини, участващи в образуването на костно-ставната система чрез създаване на разлика между отделните кости. В зависимост от вида на Синдром на Клипел-Файл има различен механизъм на наследяване, защото KFS3 KFS1 и автозомно-доминантно за KFS2 - автозомно-рецесивно.
Симптомите на синдрома Klippel-Feil
Основната трите клинични характеризиращи Синдром на Клипел-Файл, се застъпва за съкращаване на шията, изместване на линията на косата надолу по задната част на врата, нарушена подвижност на гръбначния стълб в областта на шийката на матката. Интензивността на съкращаване на шията може да се променя, и в най-тежките изпълнение ушите сключване на рамото и брадичката - на гръдната кост, затруднено преглъщане и дишане. Характерно широки лопатки разреждане и често им мазнина. Възможно е да има типично заболяване Sprengel високо положение на един от ножовете. В някои случаи, отбелязани аномалии на мускулите на раменния пояс и гънките на врата. В редки случаи, има кореновата синдром - болка, свързана с компресия на гръбначния цервикални корените.
В 50-60% от случаите, синдром на Klippel-Feil комбинира с сколиоза, в 25% от случаите - един с костни тортиколис. Може би комбинация от синдром на късо гърло с аномалии на горните крайници (полидактилия, синдактилия, вродени ампутации) крак деформации, малформации на ребра, стоматологични аномалии, лицева асиметрия, далекогледство. В 45% от пациентите, диагностицирани неправилна, аплазия или хипоплазия на бъбреците, е възможно хидронефроза, извънматочна уретер. В 25% от пациентите разкрива вродена глухота, 20% - на цепка на небцето, 15% - вродени дефекти на сърцето (отворен дуктус артериозус, VSD, ASD, dekstrapozitsiya аорта). Това може да се наблюдава или аплазия хипоплазия на белите дробове.
Нервната система е умствена изостаналост (Умствена изостаналост), епилепсия, хидроцефалия, цереброспиналната херния, микроцефалия, разстройства на движението на очите (кривогледство, птоза, Horner) синдром. От ранна възраст се характеризира с мускулна слабост в крайниците и synkineses - неволни едновременни движения на двете ръце, често само ръце. С течение на времето, може да има спастична и отпуснат пара- и тетрапрези.
Синдром Диагностика на Klippel-Feil
Проверка на диагнозата се извършва въз основа на наблюдаваните от раждането типичен триада от симптоми, данни за изследване, фамилна анамнеза, резултати от инструментални и генетични изследвания. Задайте Синдром на Клипел-Файл, с подробно описание на съществуващите аномалии, свързани възможно само в резултат на съвместната работа на много специалисти: неврология, ортопедия, генетика, кардиология, нефрология, пулмология, офталмология.
Той първо шийните прешлени радиография в 2 проекции. Когато KFS1 на рентгенографии в повечето случаи се определя от пълната синостозата 4-5 прешлените в един недиференциран конгломерат. В някои случаи, между прешлените са тънки ивици светлина, съответстваща на по-слабо развитите дисковете, което показва, че частичното синостозата че с израстването на детето да доведе до изкривяване на гръбначния стълб. Klippel-Feil синдром тип II се характеризира с комбинация от рентгенографски синостоза 7 шийните прешлени асимилация атлас и срастване на горната торакална прешлени. За да изключите KFS3 радиография на гръдни и лумбални прешлени.
По-подробна информация за костни аномалии дава CT на гръбначния стълб. Въпреки това, използването му в ранна детска възраст е ограничен поради едновременното изследване на излагане на радиация. Ако е необходимо за оценка на меки тъканни структури на засегнатата отдел (корени, гръбначния мозък) MRI на гръбначния стълб е възможно. Диференциране синдром на Klippel-Feil От вродена мускулна дистония и туберкулоза на гръбначния стълб.
Диагностика алгоритъм включва проверка на вътрешните органи: neurosonography, мозък MRI, коремна ултразвук, сърдечен ултразвук, ЕКГ, ултразвук или CT бъбречна отделителната урография, рентгенография на гръдния кош. Проведоха консултации генетика от анализа на теста на родословно дърво и ДНК.
Лечение и прогнозиране на синдром на Klippel-Feil
Извършва се предимно консервативни терапевтични мерки, насочени към предотвратяване на развитието на гръбначните изкривявания и увеличаване обхвата на движение в областта на шията. Прекарайте масаж на шийните прешлени и областта на врата, раменете и горните крайници. Препоръчителни редовни физически упражнения. Може би използването на физиотерапия. При показания Симптоматично лечение на нарушения в соматичните органи. Когато кореновата болка предписват обезболяващи средства, които носят яка Schanz.
Персистираща болка поради компресия на топ ръбове на корените е индикация за операцията. Операцията се осъществява в съответствие с нивото на техниката и представлява Bonola т. Н. tservikalizatsiyu от резекция на горните ръбове 4. Достъпът се осъществява чрез паравертебралния разрез успоредно на вътрешния ръб на острието. Операцията се извършва на 2 етапа, отделно от всяка страна.
Сама по себе си, синдром на Klippel-Feil има благоприятна прогноза от жизненоважно значение. Наличието на соматични малформации органи усложнява ситуацията и може да служи причина за преждевременна смърт. В функционално отношение, лоша прогноза, въпреки продължаващите консервативни мерки, пациентът е изразявал движенията на главата на рестрикция, степента на което зависи от вида и тежестта на синдрома. Протичането на заболяването може да се обостри срещащи се в гръбначния стълб дегенеративни промени в.
 Маточната спондилоза
Маточната спондилоза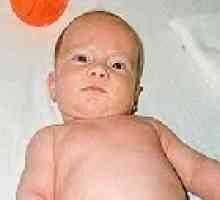 Вродена мускулна дистония
Вродена мускулна дистония Туберкулоза на гръбначния стълб
Туберкулоза на гръбначния стълб Аномалии на гръбначния стълб
Аномалии на гръбначния стълб Синостозата
Синостозата Крива шия
Крива шия Клиновидна прешлени
Клиновидна прешлени Маточната-черепната синдром
Маточната-черепната синдром Аномалия kimerli
Аномалия kimerli Синдром на гръбначния артерия
Синдром на гръбначния артерия Лечение на фрактури на C1 на прешлени
Лечение на фрактури на C1 на прешлени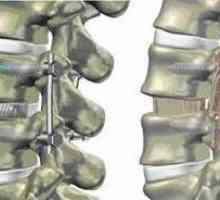 Гръбнака фиксиране в спондилолистези, намаляване на междупрешленните височината на диск
Гръбнака фиксиране в спондилолистези, намаляване на междупрешленните височината на диск Cervicalgia vertebogennaya
Cervicalgia vertebogennaya Тортиколис дете
Тортиколис дете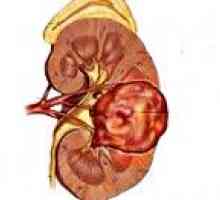 Тумор на Вилмс
Тумор на Вилмс- Загуба на слуха, увреждане на слуха
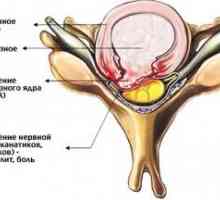 Херния шийните прешлени
Херния шийните прешлени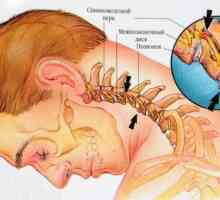 Остеохондроза на шийните прешлени
Остеохондроза на шийните прешлени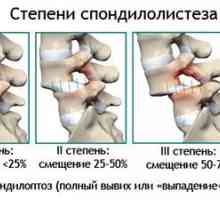 Преместването на лумбалните прешлени
Преместването на лумбалните прешлени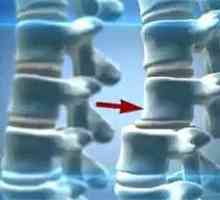 Преместването на шийните прешлени
Преместването на шийните прешлени- Синдром на Търнър
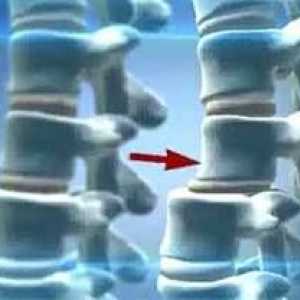 Преместването на шийните прешлени
Преместването на шийните прешлени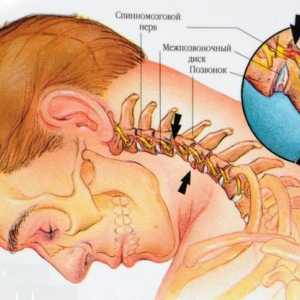 Остеохондроза на шийните прешлени
Остеохондроза на шийните прешлени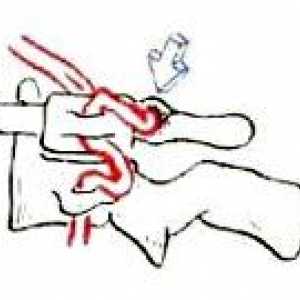 Аномалия kimerli
Аномалия kimerli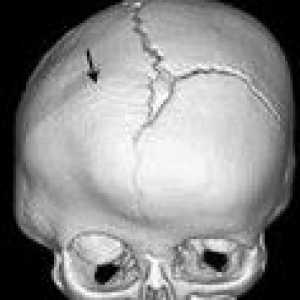 Синостозата
Синостозата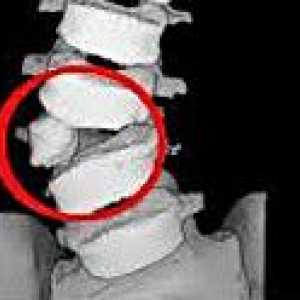 Аномалии на гръбначния стълб
Аномалии на гръбначния стълб Лечение на фрактури на C1 на прешлени
Лечение на фрактури на C1 на прешлени Cervicalgia vertebogennaya
Cervicalgia vertebogennaya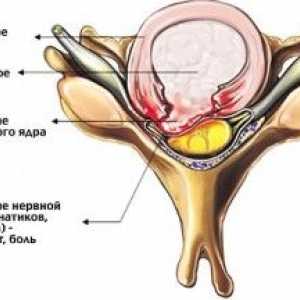 Херния шийните прешлени
Херния шийните прешлени