Machado-Joseph заболяване

Machado-Joseph заболяване
- генетично причинени спиноцеребеларна атаксия, клинична картина полиморфни комбинации от церебрална синдром със симптоми на вторичен паркинсонизъм, хиперкинеза, нарушения пирамидални под формата на спастична парализа и офталмоплегия, amyotrophy. Диагнозата се поставя въз основа на задълбочено проучване на клиничните прояви на пациента и семейството му, генеалогичен анализ, ядрено-магнитен резонанс и компютърна томография данни, идентифициране в изследването на скоростта на ДНК надвишава броя на копия на СКГ триплетния. Лечението е симптоматично. Прогнозата е лоша.Machado-Joseph заболяване

Machado-Joseph болест е описана в средата на 70-те години на 20-ти век. Предполага се, че заболяването първоначално възникнала сред жителите на Азорските острови, във връзка с която е понякога се откриват под името "Азорските острови болест." Към днешна дата, Machado-Joseph болест е широко разпространен в целия свят. дела й бяха диагностицирани при хора в САЩ, Бразилия, Япония, Индия, Европа, Китай, Австралия, Канада. Това е най-честата форма на наследствена церебрална атаксия.
В рамките на настоящата международната класификация на болестите, тази патология е потвърден като CAC Н - спиноцеребеларна атаксия тип III. Характеризира се с голяма променливост във времето на отваряне (от 10 до 70 години) и полиморфизъм на клиничните симптоми поради мултисистемна участие на церебрални и спинални структури. В зависимост от комбинацията от основни клинични синдроми отличава 3 варианти на заболяването. Полиморфизъм прояви е свързано с определени трудности при диагностиката на заболявания, които могат да бъдат приложени правилно само в тясно сътрудничество на специалисти в областта на неврологията и генетика.
Причините за заболяването Machado-Joseph
По-рано, че етиологията е неизвестна. Чрез развитието на ДНК изследвания, стана ясно, че основната субстрата действа патология генетична мутация, която се предава на поколението по автозомно доминантен начин. Аберация е локализиран в хромозома 14 (място 14q24.3-q32) и е разширяването (увеличаване на броя на повторенията) тринуклеотид свързване "цитозин-аденин-гуанин." Броят на СКГ тризнаци повтаря значително се различава и средно 62-84, а обикновено не надвишава 37. Нещо повече, проява на заболяване се среща по-рано.
Морфологично наблюдава невронална апоптоза гранулиран слой и Purkinje клетки в церебрална кора, дегенеративни промени на зъбно колело и червен ядро, субстанция нигра, моторни ядра на черепните нерви и гръбначния мозък предната рог, спиноцеребеларни пътища. В стриатума разкрива глиални пролиферация. Отличителна черта е целостта на маслините на продълговатия мозък.
Клинични варианти на заболяването Machado-Joseph
Machado-Joseph болест I тип се проявява във възрастовата периода на 10-30 години. Характеризира се с комбинация от пирамидална и екстрапирамидни симптоми. синдром на пирамидални (гръбначната лезия тракт) обикновено дебютира спастична парапареза, а след това се присъединява към слабост в ръцете, фаринкса мускулна пареза с развитието на дисфагия и дизартрия, околомоторна нерв пареза с офталмоплегия (симптом "фиксирана очи"). Има една спирка спазми, патологични рефлекси. Екстрапирамидални синдром проявява симптоми на торсионна дистония, атетоза, вторичен паркинсонизъм. Сформирана оковано бавна походка с крака широко споразумение. Маркирана нестабилност при ходене в резултат на спазми на мускулите, а не атаксия. Типични exophthalmos, големи съкращения на снопчетата език не е придружен от своята атрофия. Има фасцикулациите лицевите мускули, myokymia век. Налице е вертикална и хоризонтална нистагъм, saccades (еднопосочно движение на очите) с висока / ниска амплитуда.
Machado-Joseph заболяванията II Вид прави своя дебют в периода от 20 до 40-годишна възраст. Проявява симптоми на церебрална атаксия: Abaza, хипер- и dysmetria, нарушения в равновесието, дизартрия. Обикновено комбинацията от церебрални симптоми с пирамидални и екстрапирамидални симптоми, появяващи се в тип I. офталмоплегия и фасцикулации се наблюдава много по-често в сравнение с Machado-Joseph тип заболяване I.
Machado-Joseph заболяване тип III Тя е комбинация от церебрална атаксия и amyotrophy. Той има късното начало - след 40-годишна възраст. На фона на малкия мозък на наблюдаваните симптоми дифузна мускулна атрофия, придружен от хипотония и слабост, загуба на сухожилни рефлекси. Отличителна черта е наличието на всички видове разстройства на чувствителността на дисталния тип, което показва, полиневропатия. Proptosis лице и фасцикулации възникнат по различни източници в 20-50% от пациентите с тази форма на заболяването. Дегенерация на гръбначната тракт и повреди пирамидалната система не са типични.
Диагноза на болестта Machado-Joseph
Широкият промяната в броя на повторенията на СКГ на триплет наблюдава дори в рамките на едно и също семейство, което води до значително полиморфизъм на клиничните прояви на болестта на Machado-Joseph, свързано със значителни диагностични затруднения. Обща е наличието на различни форми на заболяването в кръвни роднини, особено когато става въпрос за различни поколения. Описва анекдотични случаи, при паркинсонизъм изиграха водеща и единствена проява на заболяването. По този начин, от ключово значение за диагнозата Machado-Joseph болест излиза Жене консултация, подробно проучване разглеждането на родословно дърво с най-много роднини на пациента, извършване на ДНК диагностика.
От гледна точка на невролог, че е важно да се идентифицират специфични разлики паркинсонов синдром, характерен за Machado-Joseph заболяване. Не са патогномонична за болестта на Паркинсон и тремор, постурална разстройства на покой и нестабилност на престояване и ходене е свързано със статичен-двигателната атаксия. Паркинсонизъм е устойчив на леводопа, въпреки че ефектът може да се наблюдава в ранните стадии на заболяването във формата на намалена мускулна ригидност.
Основни неврологични диагностика (Echo EG, ЕЕГ, REG) не дават специфични симптоми, резултатите могат да бъдат в нормални граници. CT и MRI на мозъка показва, дегенерация на малкия мозък Vermis и гуми моста. Характерна особеност е значително разширяване на IV вентрикул на фона на относителната безопасност на церебрална кора. Диференциране болестта на Machado-Joseph от други видове спиноцеребрална дегенерация, Gallervordena заболяване Spatz, оливопонтоцеребрална атрофия, атаксия на Фридрих, амиотрофична латерална склероза, атаксия, Пиер-Мари, прогресивен паралич.
Лечението и прогнозата на болестта Machado-Joseph
Една ефективна терапия е установено, все още. Симптоматично лечение. Синдромът на Паркинсон показани допаминови агонисти (Mirapex, pronoran, Пирибедил) може да се използва midantan. За отстраняване на спастичност назначен фармацевтични продукти с myorelaxant действие (Mydocalmum баклофен). производни на валпроева до-ви препоръчани за хиперкинеза (Depakinum, enkorat) или бензодиазепини (клоназепам).
За съжаление, в много случаи, симптоматично лечение не е в състояние да спре развитието на болестта Machado-Joseph. Налице е постоянна влошаване на симптомите, което води до смърт на пациента. Живот след началото на заболяването е между 10 до 20 години, в зависимост от вида на клинична патология. Най преходно изпълнение е Machado-Joseph заболяване I тип. Превенция провежда генетична консултация и пренатална диагностика в semyah- обременени от предотвратяване на раждането, като съответният генетичната мутация.
 Болест на Кьониг
Болест на Кьониг Амиотрофична латерална склероза
Амиотрофична латерална склероза Церебрална атаксия
Церебрална атаксия Neural amyotrophy на Шарко-Мари-Tooth
Neural amyotrophy на Шарко-Мари-Tooth Gallervordena заболяване Spatz
Gallervordena заболяване Spatz Хорея на Huntington
Хорея на Huntington Amyotrophy Kugelberg-Welander
Amyotrophy Kugelberg-Welander Сънна болест
Сънна болест Moya-Moya заболяване
Moya-Moya заболяване Болестта на Паркинсон
Болестта на Паркинсон Гръбначния amyotrophy
Гръбначния amyotrophy Торсионна дистония
Торсионна дистония Хипел Линдау заболяване
Хипел Линдау заболяване Екстрапирамидни синдроми
Екстрапирамидни синдроми Диагностика на рак на белия дроб
Диагностика на рак на белия дроб Болест Бинсвангер
Болест Бинсвангер- Много сперма
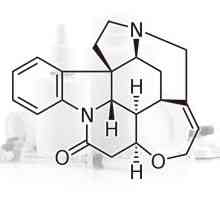 Стрихнин
Стрихнин- Урината на три порции
- Компютърна томография на бъбреците
 Магнитен резонанс (MRI). Показания, противопоказания, ядрено-магнитен резонанс
Магнитен резонанс (MRI). Показания, противопоказания, ядрено-магнитен резонанс
 Болестта на Паркинсон
Болестта на Паркинсон Статистика освободени честота на детска церебрална парализа в Русия
Статистика освободени честота на детска церебрална парализа в Русия Хорея на Huntington
Хорея на Huntington Гръбначния amyotrophy
Гръбначния amyotrophy Gallervordena заболяване Spatz
Gallervordena заболяване Spatz Екстрапирамидни синдроми
Екстрапирамидни синдроми Магнитен резонанс (MRI). Показания, противопоказания, ядрено-магнитен резонанс
Магнитен резонанс (MRI). Показания, противопоказания, ядрено-магнитен резонанс Сънна болест
Сънна болест