Синдром на Марфан

Съдържание
синдром на Марфан

Синдром на Марфан - систематично недоразвитост на съединителната тъкан в ембрионални и постнаталния период, поради структурни дефекти в колаген, и се придружава от първична лезия на мускулно-скелетната система, око, сърдечно-съдовата система. Синдром на Марфан - един от най-често срещаната наследствена kollagenopaty синдромите природа. Честотата на синдрома на Марфан на населението е ниска: Според различни автори е 1 случай на 10,000-20,000 хора, без расова и полова решителност.
Причини за възникване на синдром на Марфан
Синдром на Марфан отнася до вродени аномалии, наследствени по автозомно доминантен начин, с ясно изразен pleyotropizmom вариращи високо експресивност и пенетрантност. В основата на синдром на Марфан са мутации в FBN1 ген, отговорен за синтеза на фибрилин - основен структурен протеин на извънклетъчния матрикс, който придава еластичност и контрактилитета на съединителната тъкан. дефицит и фибрилин синдром на Марфан аномалия е вероятно да попречи на образуването на влакнести структури, намаляване на силата и еластична съединителна тъкан, невъзможността да издържат физиологични товар. Хистологичните промени са по-податливи на съдовата стена еластичен тип и лигаменти (предимно аорта и ресничните връзки на Zinn очи, съдържащи голям брой фибрилин).
синдром широк фенотипна спектър Marfan (от лека трудно различими от нормално до силно, бързо прогресиращ) се дължи на разнообразието на мутации в ген FBN1 (над 1000), и наличието на мутации в други гени (например, ген трансформиращата растежен фактор - TGFBR-2 ). Когато генетично изследване в 75% от случаите на синдром на Марфан разкрива семейството наследство, а останалите - на първичния мутация. Рискът от раждането на дете със синдром на Марфан се увеличава с възрастта на бащата (особено след като на 35).
Класификация на синдром на Марфан
В зависимост от броя на засегнатите системи излъчват няколко форми на синдром на Марфан:
- заличен - с леки промени в 1-2 системи
- изразена - с леки промени в 3 sistemah- изразени промени поне в първия sisteme- изразени промени в 2-3 или повече системи.
Тежестта на промените в синдром на Марфан може да бъде лека, умерена и тежка. Поради естеството на потока диференцират прогресивно и стабилно синдром на Марфан.
Симптомите на синдрома на Марфан
Marfan синдром се характеризира с комбиниран лезията на скелета, очите, сърдечно-съдови и нервната системни прояви на разнообразие, чрез вариране на времето на първите признаци на zabolevaniya- хронична прогресивна разбира се.
синдром на Марфан Пациентите обикновено се характеризира с висок растеж, относително кратко багажника с прекомерно дълги тънки крайници (dolichostenomelia) и паякообразни удължени пръстите (arachnodactyly) - астеничен конституция с слабо развита подкожна мастна тъкан и мускулна gipotoniey- дълга и тясна лицева скелет (dolichocephaly) - наличие на висока дъгообразна небе и захапка (Prognathism). Средната дължина на тялото при раждане в момчетата със синдром на Марфан е 53 см, крайната височина - 191 см момичета - съответно 52,5 см и 175 см.
Синдром на Марфан маркиран дисфункция на ставите (хипермобилен) - гърдите деформация (фуния или ръбести форма) гръбначния деформация (сколиоза, кифоза, kyphoscoliosis, сублуксация и дислокация на шийните прешлени, спондилолистези), както и плоско стъпало и издатината на ацетабулума.
Сърдечно-съдовите заболявания, доминираща в клиничната картина на синдром на Марфан и често определя изхода, проявява структурни дефекти като еластични стени на кръвоносни съдове, по-специално на аортата и големи клонове на белодробни артерии малформации и клапна апарат на сърдечните стени. Промени в пациенти с аортен синдром на Марфан, характеризираща се с прогресивна удължаване на възходящ част и клапан пръстен (дилатация annulo ектазия) и аневризми- лезия митралната клапа - myxomatous дегенерация клапи патологична удължаване и руптура на chordae крилото, калцификация на пръстена на клапана. Плода със синдром на Марфан може да се образува Вродени дефекти на сърцето - коарктация на аортата, стеноза на белодробната артерия, ASD и VSD. Органични и функционални промени в сърцето и кръвоносните съдове при пациенти със синдром на Марфан често са придружени от аритмии (Суправентрикуларна и камерна тахикардия, предсърдно мъждене) и развитие инфекциозен ендокардит.
форма неонатална Синдромът на най-неблагоприятната Марфан се проявява в класическия вариант при раждането, води до прогресивно сърдечна недостатъчност и смърт в първата година от живота.
За повечето случаи, синдром на Марфан се характеризира с патология на органа на зрението, включително късогледство, дислокация / сублуксация (ектопия) леща, роговицата сплескване и увеличаване на размера, хипоплазия на ириса и цилиарния мускул, кривогледство, промените в калибъра на ретинални съдове. Ектопия на лещата при синдрома на Марфан е двустранен характер на, често се развива преди навършване на 4 години и постоянно напредва, влошаване на зрителната функция.
Синдром на Марфан наблюдава увреждане на други органи и системи: нервна (.. ектазия дура вкл лумбосакрален meningocele), бронхопулмонална (спонтанен пневмоторакс, емфизема, дихателна недостатъчност), На кожата и меките тъкани (атрофичен стрии) Периодично ингвинален и бедрена херния, навяхвания и скъсани сухожилия, и извънматочна бъбреците, пикочния мехур и пролапс на матката, phlebeurysm и др.
Характерно за синдром на Марфан висока адреналин може да допринесе за постоянно възбуждане на нервите, хиперактивност, понякога развиват изключителни способности и умствени дадености.
Диагностика на Марфан Синдром
Диагнозата на синдром на Марфан основава на фамилна анамнеза, присъствие на пациент на типични диагностични характеристики от физически преглед, ЕКГ и ехокардиография, очен и радиологично изследване, молекулно генетичен анализ и лабораторни изследвания.
За диагностични критерии за синдром на Марфан вземат характерни промени в различни системи и organah- основна (голяма) от тях са: дилатация на възходящата аорта на корен / пакет и леща ектопия дурата ектазия obolochki- ръбести / фуния гърдите деформация, което изисква хирургична съотношение lecheniya- горната част на тялото към долната дължина сегмент < 0,86 или размаха рук к росту > 1,05- сколиоз (> 20 ) или спондилолистези- разширение ограничаване на лакътната става (<170о)- плоскостопие- протрузия вертлужной впадины. Остальные проявления относятся к малым критериям, а генетические (семейные) признаки – к дополнительным. Для установления диагноза синдрома Марфана необходимо наличие минимум по 1-му большому критерию в двух системах органов и 1-го малого - в третьей- в скелетной системе – присутствие минимум 4-х больших.
Също фенотипни диагностични тестове се прилагат, определяне на съотношението на четка / височина (синдром на Марфан > 11%) - средна продължителност на пръст (> 10 см) - индекс на телесна Varga на - (тегло, г / (височина) x2 - г. години / 100, следва да бъде <1,5)- тест большого пальца на арахнодактилию, тест охвата запястья.
синдром на Марфан ЕКГ за определяне на ритъма на сърцето, изразена хипертрофия miokarda- ехокардиография - откриване клапанна регургитация, увеличаване на левокамерната размери, Пролапс на митралната клапа, разгражда акорди, дилатация на аортата. На рентгенография на гръден кош може да видите на удължаването на корена и аортната дъга, увеличението на сърцето размер на CT и MRI на сърцето и кръвоносните съдове - да се идентифицират дилатация и аортна аневризма.
Аортография показано на съмнение за аневризма и аортна дисекация. Наличност ектопия на лещата уточни използване биомикроскопия и oftalmoskopii- издатина ацетабулумен метод хип radiographing настроен sustavov- дилатация дурата - гръбнака MRI.
Синдром на Марфан се определя от увеличението (2-кратно или повече) бъбречна екскреция на метаболити съединителна тъкан glyukozoaminoglikanov и техните фракции. Методът на директен автоматизирана ДНК секвениране дава възможност за идентифициране на генетични мутации в ген FBN1.
Изискване диференциална диагноза с болести, които изглежда като синдром на Марфан: хомоцистинурията, вродена контрактура arachnodactyly (синдром Beals), наследствена artrooftalmopatiey (синдром на безусловно), мас-синдром, синдром на Ehlers-Danlos, Лоис-Dietz, Shprintsena-Голдбърг семейство ектопия на лещата и други.
Марфан Синдром Лечение
Лечение и допълнително проследяване на пациенти със синдром на Марфан трябва да бъде група от специалисти: очен, кардиолог, сърдечен хирург, ортопед, генетика и интернист.
Лечение на пациенти със синдром на Марфан е насочено към предотвратяване на прогресия и развитие на усложнения, заболявания, особено в сърдечно-съдовата система. Когато диаметърът на аортата до 4 cm се назначава adrenoblokatory, калциеви антагонисти или АСЕ инхибитори. Хирургично лечение се провежда при недостатъчност на сърдечна клапа, пролапс на митралната клапа, значително разширяване (>5 cm) възходящ част и аортна дисекация. Реконструктивна хирургия на синдром на Марфан аорта, имат висок процент на постоперативна 5- и 10-годишен оцеляване. Ако е необходимо, извършване на митралната клапа. При бременни жени със синдром на Марфан и тежко сърдечно-съдово заболяване, проведено в началото хирургическа доставка чрез цезарово сечение. С оглед предотвратяването на инфекциозен ендокардит и тромбоза след хирургични процедури са предназначени антибиотици и антикоагуланти.
Marfan корекция синдром се осъществява чрез избор на очила и контактни лещи, ако е необходимо - или лазер хирургично лечение на катаракта, глаукома, премахване на разселени изкуствено имплантиране обектива. Когато изразява в скелетни смущения може да изисква хирургична стабилизация на гръбначния стълб, thoracoplasty, хип подмяната на. Също прилага kollagennormalizuyuschaya патогенетична терапия, метаболитни и витамин терапия.
Прогноза и профилактика на синдром на Марфан
Прогнози на живот при пациенти със синдром на Марфан се определя на първо място, степента на сърдечно-съдови промени, както и скелетни и очни увреждания,. Има висок риск от заболеваемост, намалява продължителността на живота (90-95% не живеят до 40-50 години) и внезапна смърт. Своевременно коригиране на сърдечно Синдром на Марфан може значително да увеличи продължителността (до 60-70 години) и да се подобри качеството на живот на пациентите.
Пациентите със синдром на Марфан трябва да бъдат под постоянно лекарско наблюдение и извършва редовна диагностика. Синдром на Марфан показва ниско до умерено ниво на физическа активност, премахва контактни спортове, спортни, изометрични упражнения, гмуркане. Жените в детеродна възраст с синдром на Марфан, трябва да се подложи на генетична консултация.
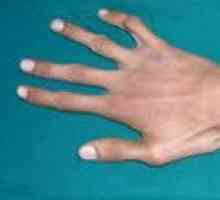 Arachnodactyly
Arachnodactyly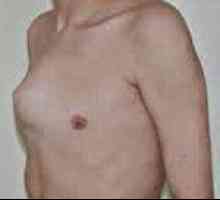 Pigeon гърдата
Pigeon гърдата Натъртване, синини, кървене в кожата
Натъртване, синини, кървене в кожата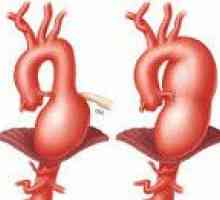 Аортна аневризма
Аортна аневризма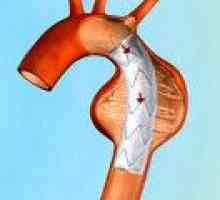 Аневризма на аортата
Аневризма на аортата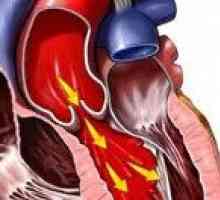 Аортна недостатъчност
Аортна недостатъчност Разрязване аортна аневризма
Разрязване аортна аневризма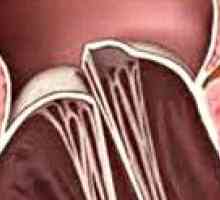 Пролапс на митралната клапа
Пролапс на митралната клапа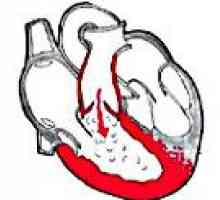 Белодробна клапан недостатъчност
Белодробна клапан недостатъчност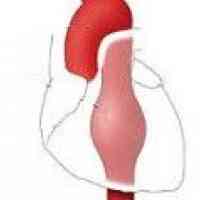 Аневризма на низходящ торакалната аорта
Аневризма на низходящ торакалната аорта Аневризми на торакалната аорта
Аневризми на торакалната аорта Лечение и профилактика на коремната аортна аневризма
Лечение и профилактика на коремната аортна аневризма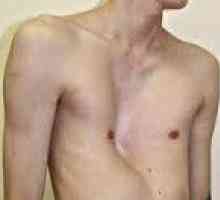 Деформацията на гръдния кош при деца
Деформацията на гръдния кош при деца Синдром на Ehlers - Danlos
Синдром на Ehlers - Danlos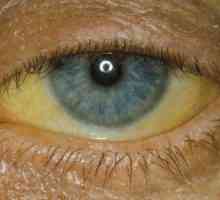 Синдром на Гилбърт
Синдром на Гилбърт Късогледство при децата
Късогледство при децата Тумори на потните жлези
Тумори на потните жлези Синдром на Съвместния хипермобилен
Синдром на Съвместния хипермобилен Съединителната тъкан дисплазия
Съединителната тъкан дисплазия Хомоцистинурията
Хомоцистинурията Синдром на Марфан
Синдром на Марфан
 Пролапс на митралната клапа
Пролапс на митралната клапа Тумори на потните жлези
Тумори на потните жлези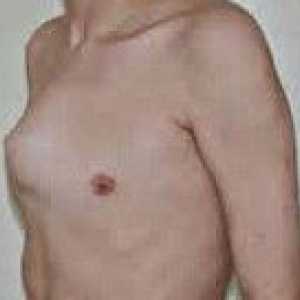 Pigeon гърдата
Pigeon гърдата Съединителната тъкан дисплазия
Съединителната тъкан дисплазия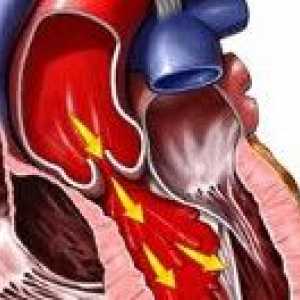 Аортна недостатъчност
Аортна недостатъчност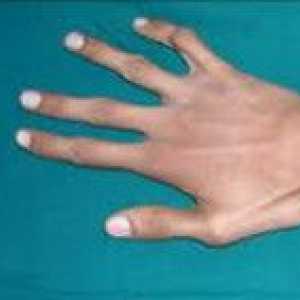 Arachnodactyly
Arachnodactyly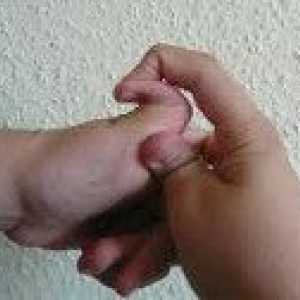 Синдром на Ehlers - Danlos
Синдром на Ehlers - Danlos Аневризми на торакалната аорта
Аневризми на торакалната аорта Деформацията на гръдния кош при деца
Деформацията на гръдния кош при деца Синдром на Съвместния хипермобилен
Синдром на Съвместния хипермобилен