Автоимунен полигландуларен синдром

Автоимунен полигландуларен синдром
Съдържание
- Автоимунен полигландуларен синдром
- Причините за автоимунен полигландуларен синдром
- Симптомите на автоимунен полигландуларен синдром тип 1
- Симптомите на автоимунен полигландуларен синдром тип 2
- Diagnosttika автоимунен полигландуларен синдром
- Лечение на автоимунен полигландуларен синдром
- Прогнозиране на автоимунен полигландуларен синдром
Автоимунен полигландуларен синдром

Автоимунен полигландуларен синдром (APGS) - immunoendokrinnoe разстройство се характеризира с основна функционално увреждане на няколко жлези с вътрешна секреция, и не ендокринни орган-специфични заболявания. В ендокринология разграничи автоимунен полигландуларен синдром тип 1 и 2 (APGS 1, 2), които имат техните генетични, имунологични и клинични характеристики.
Автоимунните жлезите с вътрешна секреция синдром тип 1 обикновено се проявява в детството (10-12 години) и понякога се нарича в литературата с термина "младежката poliendokrinopatiya семейство." Разпространението на APGS-1 е малка, но болестта е малко по-често при мъжкото население, най-вече жителите на Финландия, Сардиния, Иран, поради дългосрочния генетична изолация на тези народи. Автоимунен полигландуларен синдром тип 1, съдържа триада от симптоми: надбъбречна недостатъчност, gipoparatiroz и кандидос.
Най-често срещаният вариант на множество автоимунни ендокринопатии е автоимунен синдром на жлезите тип 2, който се развива при възрастни (на възраст 20-30 години) - жени преобладават сред пациентите. APGS компоненти са 2-надбъбречна недостатъчност, захарен диабет Тип 1, автоимунен участие на щитовидната жлеза по вид на първичен хипотиреоидизъм или хипертиреоидизъм.
В допълнение към ендокринни заболявания, автоимунни полигландуларен синдром тип 1 и 2 придружени от други орган-специфични проявления.
Причините за автоимунен полигландуларен синдром
И двата вида автоимунен полигландуларен синдром са генетично определени, както е посочено от семейството и наследствени заболявания. Така APGS-1 засегнати братя и сестри на един generation- автоимунна жлезите с вътрешна секреция синдром тип 2 - представители на едно и също семейство в продължение на няколко поколения.
Автоимунен полигландуларен синдром тип 1 - единственият известен автоимунно заболяване като моногенна в природата. За да се развие APGS-1 генна мутация води автоимунно регулатор (AIRE), разположен на дългото рамо на хромозома 21 (21q22.3). Автоимунен полигландуларен синдром-1 се наследява по автозомно рецесивен начин и не се свързва с HLA хаплотипа.
Автоимунен полигландуларен синдром тип 2 се свързва с HLA-хаплотипа (антигени DR3, DR4, DR5, В8, DW3). Много е вероятно, че механизмът на развитие APGS-2 е свързано с анормална експресия на антигени HLA-система на клетъчните мембрани на жлезите с вътрешна секреция, а именно под влиянието на външни фактори.
Симптомите на автоимунен полигландуларен синдром тип 1
Автоимунен полигландуларен синдром тип 1 обикновено първо се проявява в първите 10 години от живота, развитието на грануломатозен кандидоза (кандидоза на кожата и лигавиците) и хипопаратироидизъм. В бъдеще (често десетилетия), свързани хронична бъбречна недостатъчност (болест на Адисон). Клиничните критерии за диагностика на синдрома е комбинация от две от трите споменати ендокринни разстройства.
Кандидоза на APGS-1 се генерализирани и се отразява на лигавицата на устата, гениталиите, дихателните пътища и стомашно-чревния тракт и кожата, ноктите ролки, ноктите. Хипопаратиреоидизъм се проявява конвулсии на мускулите, кожата парестезия, laringospazmom, припадъци, напомнящ епилепсия. Надбъбречна недостатъчност при пациенти с автоимунни полижлезиста синдром увеличава скрити и могат да бъдат показани за първи път на фона на криза на addisonicheskim стресово състояние.
Класическата триада на автоимунна жлезите с вътрешна секреция синдром често се свързва с първичен хипогонадизъм (45%) алопеция (30%) синдром на малабсорбция (23%) злокачествена анемия (14%) хроничен автоимунен хепатит (12%) автоимунна тиреоидит (10%), инсулин-зависим диабет (4%) витилиго (4%). По-рядко срещани дистрофия на нокътната плочка, хипоплазия на емайла, гломерулонефрит, бронхиална астма, катаракт.
На фона на автоимунна жлезите с вътрешна секреция синдром тип 1 при жените отпразнува яйчниците хипоплазия (Автоимунен оофорит), придружен от първичен или вторичен аменорея. Мъж хипогонадизъм се проявява намалено либидо, импотентност, безплодие.
Симптомите на автоимунен полигландуларен синдром тип 2
Проявите на автоимунни 2 сметки на жлезите с вътрешна тип синдром на зряла възраст (около 30 години). Първата проява ендокринопатия обикновено хронична надбъбречна недостатъчност. Други автоимунни компоненти (диабет тип 1, автоимунен тироидит), са склонни да се приведе в 7-10 години или повече.
Пациенти с автоимунна жлезите с вътрешна секреция синдром тип 2 често развиват първичен хипогонадизъм, миастения, витилиго дерматит, алопеция, автоимунен гастрит, цьолиакия, стеаторея, poliserozita (плеврит, перикардит, асцит) тимома. може да се получи автоимунен полигландуларен синдром атрофия на зрителния нерв, тумор на хипофизата, автоимунна тромбоцитопенична пурпура и т. г.
Най-често срещани в практиката версия на автоимунен полигландуларен синдром тип 2, който съчетава първичен хронична надбъбречна недостатъчност с автоимунни тироидни заболявания клинично: автоимунен тиреоидит, най-малко - дифузен токсичен гуша (Синдром на Шмит).
Множествена ендокринопатия взаимно влоши помежду си и да влоши протичането на заболяването.
Diagnosttika автоимунен полигландуларен синдром
Критерии за клинична диагноза лабораторни и се потвърждават от инструментални изолирани компоненти автоимунен полигландуларен синдром (кожно-лигавична кандидоза и хипопаратироидизъм в APGS HNN HNN-1, автоимунен тиреоидит и с диабет APGS-2). Когато APGS тип 1 държи най-значително молекулно генетичен анализ за идентифициране на характеристика генна мутация.
Лабораторни изследвания автоимунен полигландуларен синдром включва определяне биохимичните кръвни показатели (ниво на общ и йонизиран калций, фосфор, калий, натрий, билирубин, трансаминази, алкална фосфатаза, общ протеин, уреа, креатинин, глюкоза), CBS кръв и др. Важно диагностична информация даде изследвания на хормони: TTG, свободен тироксин, паратироиден хормон, АСТН, инсулин, с-пептид, кортизол, ренин, алдостерон, соматомедин с, TPO, антитела към бета клетките на панкреаса и GAD, testoster това, LH, FSH и други.
Инструментални изследвания включват коремна ехография, щитовидната жлеза, тазови органи (при жените) и скротума при мъжете, ехокардиография, CT надбъбречната жлеза. Когато това е необходимо консултирани специалисти - Диабетолог, гастроентерология, хепатология, дерматолог, mycologist, невролог, хематолог, офталмолог, гинеколог, ендокринолог, андролог, ревматолог.
Лечение на автоимунен полигландуларен синдром
Лечението на автоимунни полигландуларен синдром е сложна задача и се състои от лечението на отделните компоненти. В основата на патогенетична терапия на постоянен хормон заместваща терапия при функционални засегнати недостатъчност ендокринни жлези. Когато надбъбречна недостатъчност, определени глюкокортикоиди (хидрокортизон, дексаметазон, преднизолон, триамцинолон), минералокортикоиди (Doxey, триметилацетат деоксикортикостерон и др.), В хипотиреоидизъм - L-тироксин.
Когато се използват кандидоза придружаващия автоимунен полигландуларен синдром тип 1 антимикотици. При диабет срещу автоимунен полигландуларен синдром тип 2 могат да изискват имуносупресивна терапия с циклоспорин.
Пациентите се препоръчва да се използват повишени количества аскорбинова киселина и техни соли. Забранени употреба на алкохол, някои лекарства.
Прогнозиране на автоимунен полигландуларен синдром
Ранното откриване на автоимунен полигландуларен синдром и провеждане подходяща заместваща терапия може да контролира заболяването. Въпреки това, способността да се работи обикновено намалява - пациенти възложени II-III увреждане.
Пациенти с автоимунна жлезите с вътрешна секреция синдром подлежи на диспансерно наблюдение ендокринолог и други специалисти. В условията на стрес, успоредно протичащи инфекции, пациенти физическо или умствено натоварване трябва да се увеличи дозата на хормони. Необходимост от незабавно лечение на лекар за всяко влошаване на здравето. Летален изход на автоимунен полигландуларен синдром може да възникне от ларингоспазъм, висцерална кандидоза, остра надбъбречна недостатъчност.
 Ахлорхидрия
Ахлорхидрия Автоимунен хепатит
Автоимунен хепатит Хроничен атрофичен гастрит
Хроничен атрофичен гастрит Хроничен вирусен хепатит
Хроничен вирусен хепатит Хроничен гастрит
Хроничен гастрит Първична билиарна цироза
Първична билиарна цироза Заболявания на щитовидната жлеза при жените и мъжете
Заболявания на щитовидната жлеза при жените и мъжете Загуба на слуха, увреждане на слуха
Загуба на слуха, увреждане на слуха Тиреоидит
Тиреоидит Тиреоидит на Хашимото
Тиреоидит на Хашимото Тиреоидит на Хашимото
Тиреоидит на Хашимото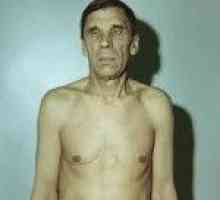 Panhypopituitarism
Panhypopituitarism Лимфом на щитовидната жлеза
Лимфом на щитовидната жлеза- Тиреоидит
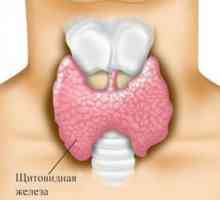 Автоимунните тиреоидит (тиреоидит на Хашимото)
Автоимунните тиреоидит (тиреоидит на Хашимото)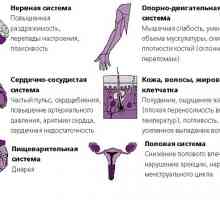 Тиреотоксикоза (хипертиреоидизъм)
Тиреотоксикоза (хипертиреоидизъм)- Хипотиреоидизъм
- Плазмафереза
 Автоимунни заболявания
Автоимунни заболявания Общо протеин в кръвта
Общо протеин в кръвта Бременност с автоимунно заболяване на черния дроб (хепатит, холангит, цироза)
Бременност с автоимунно заболяване на черния дроб (хепатит, холангит, цироза)
 Ахлорхидрия
Ахлорхидрия Заболявания на щитовидната жлеза при жените и мъжете
Заболявания на щитовидната жлеза при жените и мъжете Тиреотоксикоза (хипертиреоидизъм)
Тиреотоксикоза (хипертиреоидизъм) Тиреоидит
Тиреоидит Автоимунни заболявания
Автоимунни заболявания Тиреоидит на Хашимото
Тиреоидит на Хашимото Хроничен гастрит
Хроничен гастрит Първична билиарна цироза
Първична билиарна цироза Тиреоидит на Хашимото
Тиреоидит на Хашимото